
If you’ve looked at this blog for any length of time — then of course you know that multi-component reactions and cool heterocycles are dear to me….but reading through this article (ARKIVOC 2012) caught two things that interest me on a different level: solventless reactions (where is all the data) and replacements of reagents and metals that are always tried because we don’t think as much as we do. Maybe it is just because I made a coupla’ hundred of these — well to protect the innocent and guilty, I had a number of modifications on the aldehyde and isocyanide (several metals are used in this strategy as well — Groebke reaction).
AS a starter — it is not hard to come across an article that uses a clay or solid acidic resin to do some solventless microwave chemistry. There a place for it in a so many areas — the problem stems from the variability of the reactions — sometimes I read through these like I am being graded for my 6th grade writing assignment (you remember, who are they? skipping theme content? and on and on. Well, not this paper, but when I started to read it, that’s the thought that came to me because so many papers doing this repeat a theme: we are using the $100 microwave with a reaction in an open beaker with clay….and it goes to 180C in near quantitative yield — really? Come on, I bet if you moved it around the cavity it would give you a different yield — like I said I have learned to expect this….and it’s why when we try, we are sometimes off so much on something that it doesn’t repeat at all….thanks, right?

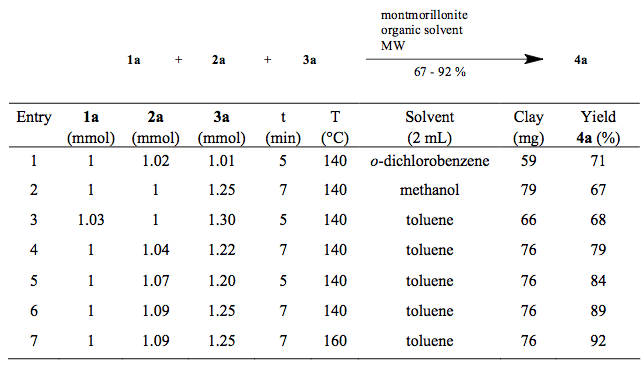
3-component amino pyridine, iscoyanide and aldhyde mw with clay
With some slight change in the stoichiometry and a few changes in solvent, we find that higher temperature and toluene provide about a 20% increase in yield (compared to a lower oil bath yield)— I am not surprised — stirring, who knows if the clay has a limit in heating or we simply haven’t gone to high enough temperature (maybe that is a function of the vessel, but I think we should have gone higher — OK well 7 min is a pretty short reaction — I give them kudos!
Moving from solventless to organic solvents
Now this really got me going — maybe there is some likemindedness here….moving from organic solvents to ionic liquids — higher heating capacity than the clay, and we can still agitate the reaction if needed. Well well, not all IL processes are proved true. Depending on which was used, decomposition and low yields are the story. But there were a couple of strong reactions to note — don’t you love the heating capacity of ILs — almost no power added provided pretty decent temperatures — but it is interesting that the times went up significantly — maybe there is competition between the clay and IL for the reactants?

Same reaction with ILs
You will have to dig into the article for yourself, because there wasn’t much of a transition from ILs to guanidinium sustrates to replace the ILs and then the clay itself. Ionic indeed, about the same or a little better than clay, Entry 5c as the best tested. Maybe there are some good salt substitutions for this reaction after all. This article made me think a bit on how I would use the information — plenty of things to try. Happy Reading!

again, with guanidinium salts

best from the list tried
















